Interrupción de arco aórtico
Descargar PDFDefinición:
De la porción transversa de la aorta normal emergen 3 grandes arterias que nutren la cabeza y los brazos: el tronco arterial braquiocefálico o arteria innominada (que dará las arterias subclavia derecha y carótida derecha), la arteria carótida izquierda y la arteria subclavia izquierda, en ese orden (Ver Anatomía cardíaca en Corazón normal).
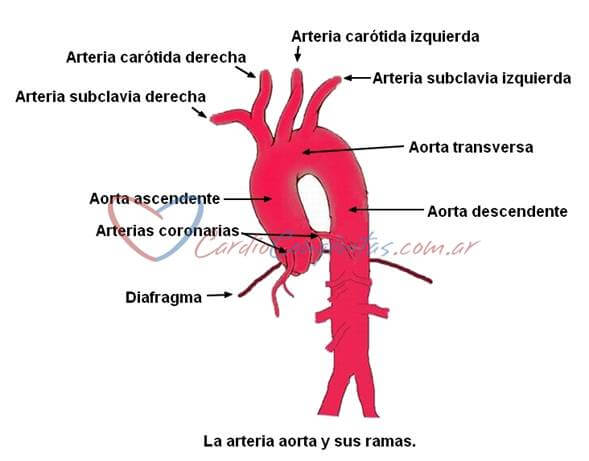
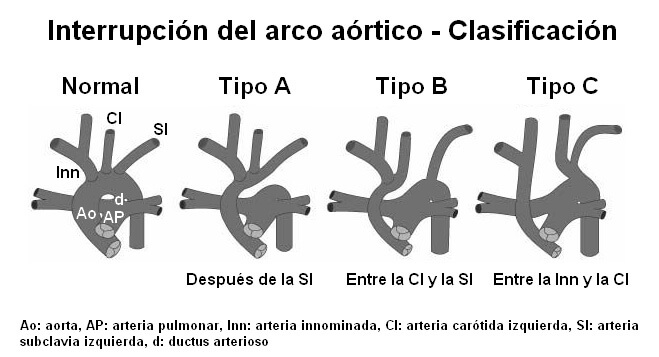
La interrupción del arco aórtico es una malformación congénita rara, caracterizada por la discontinuidad anatómica entre los segmentos del arco aórtico.
La aorta ascendente y algunas ramas de la aorta transversa quedan conectadas al ventrículo izquierdo, mientras que las ramas restantes de la aorta transversa y la aorta descendente quedan unidas al ductus arterioso y por lo tanto a la arteria pulmonar y el ventrículo derecho. De acuerdo al sitio en donde se encuentre interrumpida la aorta se definen 3 subtipos de interrupción: tipos A, B y C. El más frecuente es el tipo B (60%). Le siguen el tipo A (39%) y el tipo C (1%).
En la gran mayoría de los casos, existe otro defecto anatómico asociado a esta interrupción de la aorta, que es la presencia de una comunicación interventricular (CIV). Esto consiste en la presencia de un agujero que comunica ambos ventrículos entre sí, permitiendo el pasaje de sangre en forma anormal desde un lado al otro del corazón.
Otras malformaciones asociadas son la arteria subclavia derecha aberrante y la estenosis subaórtica. La primera de ellas consiste en la ubicación anormal de la arteria subclavia derecha, que en lugar de emerger de la arteria innominada, lo hace sola desde la aorta y como última rama, después de la arteria subclavia izquierda. Para poder llegar desde ese nacimiento hasta el brazo derecho, debe cruzar por detrás del esófago, por lo que a veces produce cierto grado de obstrucción esofágica con síntomas digestivos tales como vómitos.
La estenosis subaórtica consiste en la disminución del diámetro (con la consiguiente obstrucción al flujo de sangre) de la salida del ventrículo izquierdo, justo antes del nacimiento de la arteria aorta. Esta malformación tiene importantes implicancias en la elección de la cirugía más conveniente para el paciente, como veremos más adelante.
Presentación clínica:
En el recién nacido con interrupción del arco aórtico, la irrigación sanguínea de la mitad inferior del cuerpo depende del ductus arterioso. Mientras éste permanezca abierto, el bebé estará estable clínicamente. Sin embargo, su cierre normal días después del nacimiento desencadena una falla cardíaca severa (Ver Manifestaciones clínicas en Diagnóstico y tratamiento) y un shock con alto riesgo para la vida si no es tratado en forma inmediata con prostaglandinas para abrir el ductus y medidas agresivas tales como diuréticos, inotrópicos y asistencia respiratoria mecánica. En raras ocasiones puede observarse una coloración diferente entre la mitad superior y la inferior del cuerpo, siendo más azulada en esta última.
Diagnóstico:
La sospecha diagnóstica surge ante un bebé con insuficiencia cardíaca en los primeros días de vida (Ver Diagnóstico en Diagnóstico y tratamiento). La diferencia de pulsos entre los 4 miembros puede orientar a la localización de la interrupción. La radiografía de tórax muestra severa cardiomegalia (agrandamiento del corazón) y exceso de sangre en los pulmones.
El ecocardiograma es el método diagnóstico de excelencia para estos casos. Define la anatomía del arco aórtico, la anatomía intracardíaca (presencia y ubicación de la comunicación interventricular y otros defectos) y detecta la estenosis subaórtica.
Dada la utilidad de la ecocardiografía, es raro que deban realizarse otros estudios complementarios tales como el cateterismo, la tomografía computada o la resonancia magnética nuclear. Estos se reservan para casos puntuales en los que el cuadro no queda completamente claro.
Tratamiento:
El tratamiento inicial debe estar orientado a compensar clínicamente al paciente. Esto incluye la conexión del paciente a un respirador y la administración de medicación endovenosa (prostaglandinas para abrir el ductus, inotrópicos para mejorar la función cardíaca y diuréticos para disminuir la cantidad de líquido en los pulmones).
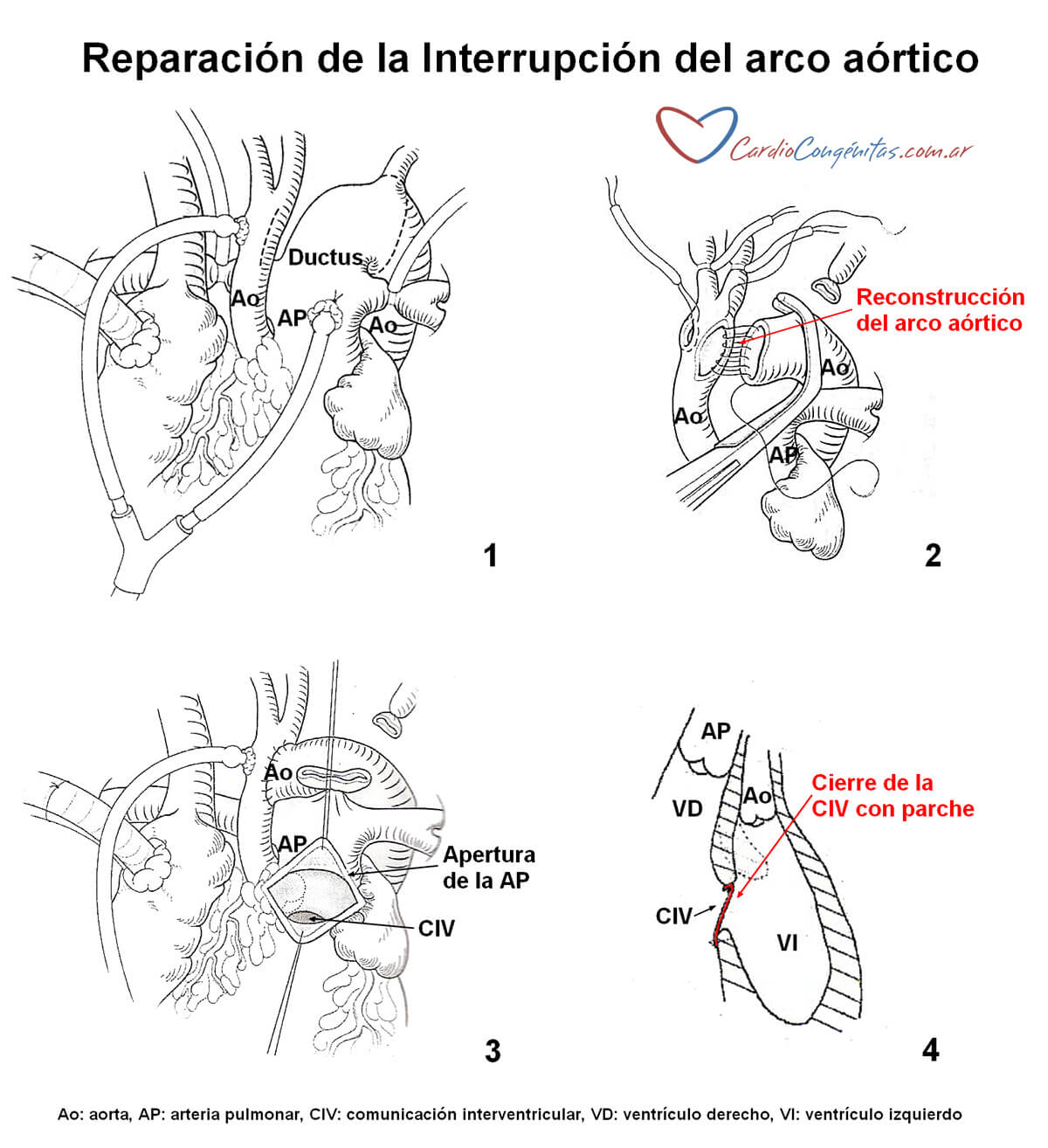
Una vez compensado, se debe llevar al paciente a quirófano para practicarle una cirugía correctora (Ver Cirugía en Diagnóstico y tratamiento). Antiguamente, solía practicarse la reparación en dos etapas. La primera consistía en la reparación del arco aórtico y el cierre parcial de la arteria pulmonar (cerclaje) abordando el tórax por debajo del omóplato izquierdo, sin circulación extracorpórea. En una segunda cirugía se abordaba el tórax por el medio, se sacaba el cerclaje de la arteria pulmonar y se cerraba la comunicación interventricular usando circulación extracorpórea.
Actualmente, la tendencia mundial es a corregir todo en una sola cirugía abordando el tórax por el medio a través del esternón. Una cuestión crítica para la elección de la cirugía es determinar si una vez operado la salida del ventrículo izquierdo quedará chica, defecto denominado estenosis subaórtica. Si bien no existe un valor claro de corte para separar a aquellos pacientes que la desarrollarán de los que no lo harán, en general se puede asumir que si el diámetro (medido en milímetros) de la salida del ventrículo izquierdo es menor al valor del peso del paciente, la obstrucción izquierda postoperatoria será importante. Por el contrario, si el diámetro es mayor al peso + 2 mm probablemente no exista obstrucción postoperatoria. Como ejemplo pongamos a un bebé de 3 kg. Si la salida del ventrículo es mayor a 5 mm probablemente no exista obstrucción postoperatoria, pero si es menor a 3 mm, seguro la desarrollará. Los valores intermedios representan un gran desafío para el equipo médico y deberán ser evaluados puntualmente.
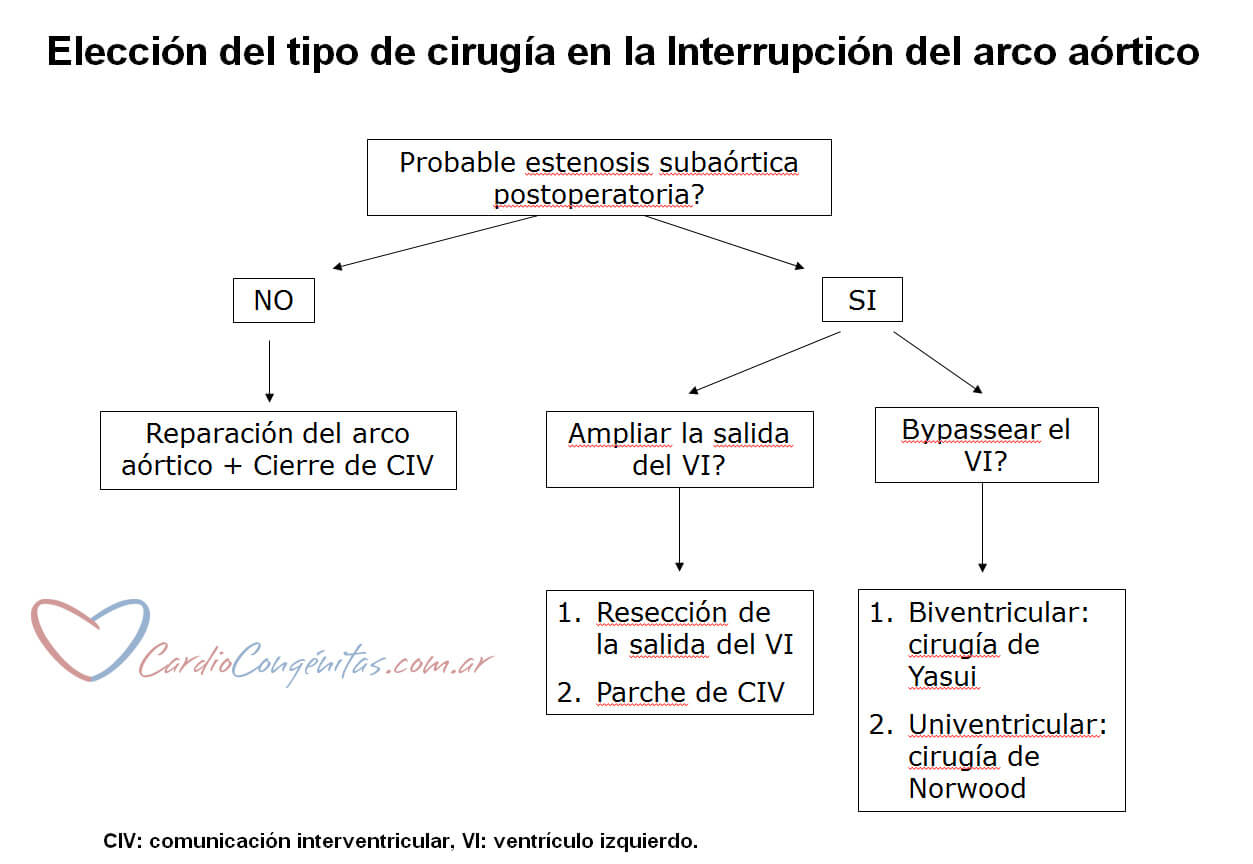
Si la obstrucción es poco probable lo que se hará será reparar el arco aórtico y cerrar la comunicación interventricular.
Si la obstrucción es muy probable luego de la corrección, existen 2 estrategias posibles. La primera es ampliar la salida del ventrículo izquierdo resecando el músculo que la obstruye y colocando el parche de la comunicación interventricular de una manera tal que traccione de los tejidos y libere la salida del ventrículo.
La segunda estrategia consiste en saltear la salida del ventrículo izquierdo. En esta situación tenemos a su vez 2 opciones. Si el ventrículo izquierdo es muy chico y no se considera viable, se realizará la cirugía de Norwood (Ver Hipoplasia de corazón izquierdo en Cardiopatías congénitas). En esta cirugía se reconstruye la aorta y se la une a la arteria pulmonar de manera que ambos ventrículos envían su sangre hacia todo el organismo. Como no existe otra fuente de sangre a los pulmones, se agrega una anastomosis sistémico-pulmonar (una conexión entre una rama de la aorta y una de las ramas de la arteria pulmonar) o un tubo angosto desde el ventrículo hacia las ramas de la arteria pulmonar (cirugía de Sano).
En el caso de que el ventrículo izquierdo sea de adecuado tamaño se podrá intentar la reparación biventricular mediante la cirugía de Yasui, en la que se reconstruye la aorta como en la cirugía de Norwood, se tuneliza la sangre del ventrículo izquierdo hacia la salida de la arteria pulmonar con un parche a través de la comunicación interventricular y por último se conecta el ventrículo derecho con las ramas de la arteria pulmonar con un tubo, de manera de aportarle sangre a los pulmones desde el ventrículo. Este tipo de cirugía es muy compleja y de alto riesgo, de manera que en muy pocas ocasiones se la realiza.
Pronóstico:
El primer desafío consiste en superar la cirugía y el postoperatorio. Ambos representan un riesgo importante para la vida del paciente, que habitualmente permanece en terapia intensiva muchos días, conectado a respirador y con drogas de todo tipo (Ver Postoperatorio en Diagnóstico y tratamiento). Si no existen defectos residuales tales como obstrucción del arco aórtico reconstruido, comunicación interventricular o estenosis subaórtica, en general el pronóstico es favorable, con una mortalidad cercana al 5% en los centros de mayor experiencia. En ausencia de esos defectos residuales, la posibilidad de tener que realizar otra cirugía es muy baja y la calidad de vida del niño será excelente .